










Post Relacionados


Cómo minimizar los errores en la espectrometría de masas de reticulación
2025-09-23La espectrometría de masas de reticulación (XL-MS) ha impulsado estudios internacionales históricos, desde el mapeo integrador del complejo de poros nucleares humanos hasta los análisis en toda la célula de todo el proteoma utilizando enlazadores de escisión de MS como DSSO, y el trabajo rápido de interacción de la proteína SARS-CoV-2. Las aplicaciones abarcan biología estructural y modelado integrador (restricciones para crio-EM / NMR), interactómica nativa, dinámica conformacional cuantitativa (QCLMS), arquitectura de cromatina / RNP, mapeo de epítopos e interfaces para productos biológicos y estudios de mecanismos de acción en virología y descubrimiento de fármacos. XL-MS complementa los métodos clásicos al capturar las restricciones de distancia en solución e incluso in situ, lo que permite una visión estructural a nivel de sistemas.
(Mecanismo molecular de interacción entre el SARS-CoV-2
Y las células huésped y la terapia intervencionista | Transducción de señales y terapia dirigida)
La espectrometría de masas de reticulación (XL-MS) es una forma poderosa de trazar las interacciones proteína-proteína, sin embargo, muchos laboratorios aún lidian con falsos positivos que desdibujan la biología real y toman decisiones lentas. En Longlight Technology, abordamos el problema en su origen: química estricta, preparación repetible y análisis transparente, para que pueda confiar en los mapas que construye.
1) Por qué ocurren los falsos positivos – y cómo se infiltran
Los falsos positivos rara vez comienzan en la etapa de software. Comienzan en el banquillo. La reticulación excesiva empuja a las proteínas a emparejamientos no fisiológicos. El enfriamiento insuficiente deja grupos reactivos que siguen vinculándose mientras maneja la muestra. La digestión enzimática apresurada o inconsistente crea especies peptídicas que se disfrazan de enlaces cruzados reales. Agregue matrices complejas y los globos espaciales de búsqueda, haciendo que los espectros limítrofes parezcan convincentes.
La realidad del día a día agrega más riesgo. Un tampón preparado ligeramente apagado. Un enfriamiento que dura 5 minutos. Un lote de enzimas diferente. Ninguno de estos pasos en falso parece dramático por sí solo, pero juntos empujan a la espectrometría de masas de reticulación hacia el error. El resultado es un conjunto de datos que "parece" rico pero que exige horas de triaje para encontrar lo que realmente importa.
A pesar de estas trampas, XL-MS tiene fortalezas únicas que no desea mitigar. La reticulación química junto con la espectrometría de masas estabiliza los complejos antes del análisis, preserva las interacciones débiles o de corta duración y proporciona pistas espaciales sobre la disposición y la conectividad de las proteínas. Es compatible con la proteómica estructural, se puede realizar en células y no requiere etiquetas especiales. El objetivo no es ralentizar el método, sino eliminar el ruido sin perder velocidad.
✅ Puntos débiles comunes que vemos
• Enlaces cruzados no específicos formados durante la preparación de la muestra
• Reticulación de baja abundancia enterrada por péptidos abundantes
• Digestión incompleta o desigual que genera artefactos
• Búsquedas amplias combinadas con filtros indulgentes en el análisis de datos
Estos problemas se pueden resolver con una vista del sistema en lugar de una solución de un solo paso.
2) Luz larga‘para reducir los falsos positivos
Nuestra perspectiva es simple: tratar la espectrometría de masas reticulada como una cadena conectada – química, preparación, detección e interpretación. Diseñamos el plan de reticulación en torno a su pregunta biológica y tipo de muestra, luego mantenemos las condiciones estrictas, documentadas y repetibles.
✅ Enriquecimiento que RAflojar Signal Unbuey NOise
Un péptido reticulado es raro por definición. Enfatizamos el enriquecimiento selectivo después de la digestión para aumentar su proporción antes de que el instrumento vea la muestra. Las entradas más limpias reducen las identificaciones ambiguas y reducen su tasa efectiva de falsos descubrimientos. El efecto es práctico: mayor confianza con menos rescates manuales río abajo.
✅ Cinco Habits que CExaminar en Cmás delgado Data
• Comience con un plan de reticulación basado en preguntas, no con un protocolo único
• Controle la ventana de enlace, temperatura y enfriamiento con la misma disciplina en cada ejecución
• Estandarizar la digestión y la limpieza para limitar los parecidos a los péptidos
• Utilizar el enriquecimiento dirigido para aumentar el rendimiento de péptidos reticulados antes de la EM
• Establecer y documentar umbrales de análisis vinculados a los objetivos del estudio, luego cumplirlos
Esta disciplina conserva las condiciones de victoria de XL-MS: alto rendimiento, aplicabilidad intracelular y sin etiquetado especial. Cuando una llamada de estructura es importante, recomendamos combinar la espectrometría de masas de reticulación con métodos ortogonales como la crio-EM o la cristalografía de rayos X. La combinación reduce el riesgo de interpretación de cualquier tecnología y ayuda a crear modelos que su equipo puede defender.
Qué “PPráctico“ Looks como en Ynuestro Lapagado
No le pedimos que cambie su ciencia, solo que bloquee las variables que importan. Obtiene un plan con marca de tiempo, verificaciones de reactivos y una breve lista de puntos de control que los técnicos pueden seguir. El resultado no son solo datos más limpios; Es un proceso que puede repetir el próximo trimestre con nuevas muestras y la misma confianza.
CTA: ¿Quieres menos falsos positivos y mapas de interacción más nítidos? Hable con Longlight Technology sobre un plan XL-MS basado en preguntas que se adapte a sus muestras y plazos.
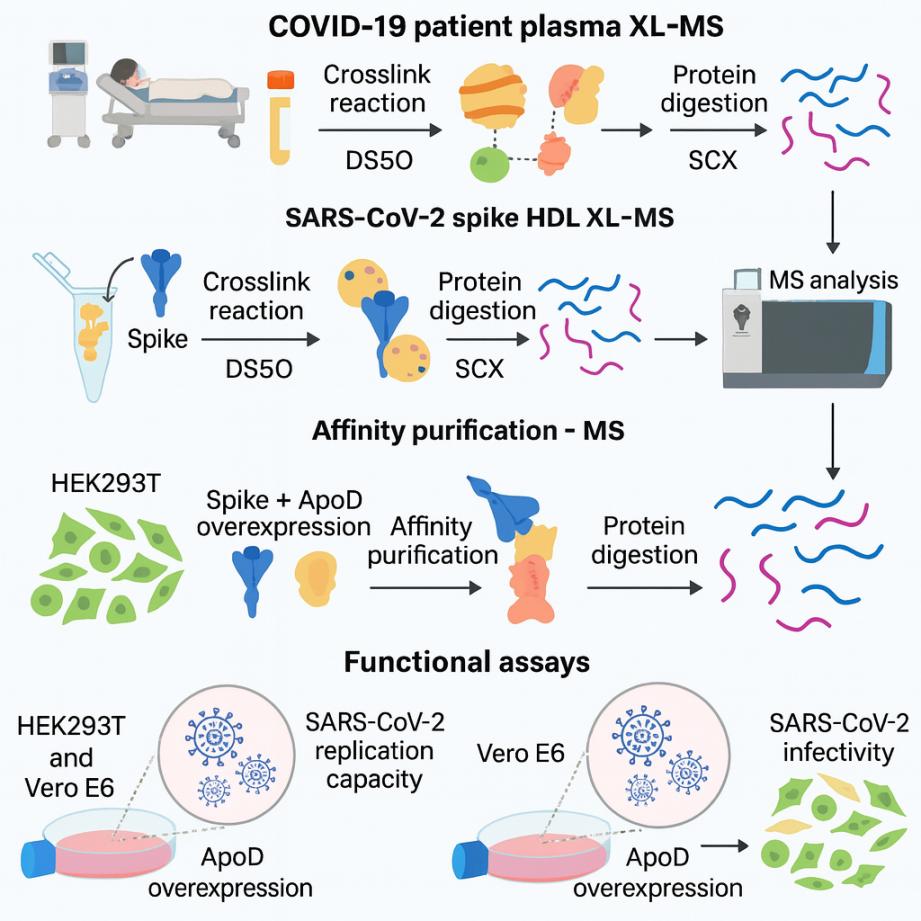
(La espectrometría de masas reticulada descubre interacciones entre lipoproteínas de alta densidad
y la glicoproteína de pico del SARS-CoV-2 – ScienceDirect)
3) De la muestra al informe: un servicio XL-MS de extremo a extremo que puede auditar
Puede enviar material pre-reticulado, o le ayudaremos a diseñar la estrategia de reticulación y luego recibiremos las muestras. A partir de ahí, nuestro equipo ejecuta la digestión enzimática, el enriquecimiento selectivo, la detección de espectrometría de masas de reticulación y el análisis de datos estructurados. El informe final muestra redes de interacción y sitios de enlaces cruzados con métodos claros, para que su equipo pueda repetir, escalar o integrarse con otros estudios.
Qué Yo RRecibe, STEP por STEP
Planificación → estrategia de reticulación → digestión → enriquecimiento → detección → interpretación. Cada paso incluye puntos de control y notas. Si algo se desvía, podemos ver dónde y por qué. Esa trazabilidad reduce la ambigüedad y mantiene constante el ritmo de descubrimiento sin necesidad de apagar incendios constantemente.
Nuestra oferta XL-MS se encuentra dentro de un ecosistema de productos y servicios más amplio diseñado para que su laboratorio siga avanzando. Apoyamos la genómica y la proteómica modernas con instrumentos avanzados, reactivos confiables y consumibles adecuados para su propósito. Esa continuidad es importante: cuantas menos transferencias haya entre proveedores, más fácil será preservar la calidad en todos los proyectos.
Más allá de la espectrometría de masas de reticulación, permitimos aplicaciones complementarias que muchos equipos ejecutan en paralelo. Para preguntas sobre la cromatina de proteínas, apoyamos ChIP-seq, que combina la inmunoprecipitación de cromatina con la secuenciación de próxima generación para identificar sitios genómicos unidos por factores de transcripción específicos o histonas. Para los flujos de trabajo diarios, proporcionamos consumibles y kits (geles de agarosa prefabricados, eliminadores de ácidos nucleicos, tubos Qubit, kits de extracción de ácidos nucleicos y kits de preparación de bibliotecas) para que pueda mantener estándares consistentes desde la toma de muestras hasta la entrega de datos.
Por qué TEAMS CTecnología de luz larga
• Un único socio para la química, el diseño del flujo de trabajo y el análisis XL-MS
• Métodos claros y auditables que se escalan desde el piloto hasta la producción
• Instrumentos, reactivos y consumibles que respaldan la genómica y la proteómica bajo un mismo techo
La recompensa no es abstracta. Menos falsos positivos significan decisiones más rápidas, cifras más creíbles y menos tiempo dedicado a discutir con sus propios datos. Sus científicos pueden concentrarse en la biología, no en el triaje. Sus partes interesadas ven progresos, no advertencias. Y sus modelos se trasladan al siguiente programa con menos ediciones y pruebas más sólidas.
CTA: ¿Listo para apretar su tubería XL-MS y reducir el ruido en la fuente? Póngase en contacto con Longlight Technology para programar una breve consulta. Describiremos un flujo de trabajo de enriquecimiento de péptidos reticulados que protege la especificidad, acelera el análisis y hace que sea más fácil confiar en sus datos de espectrometría de masas de reticulación, ejecución tras ejecución.