










Post Relacionados
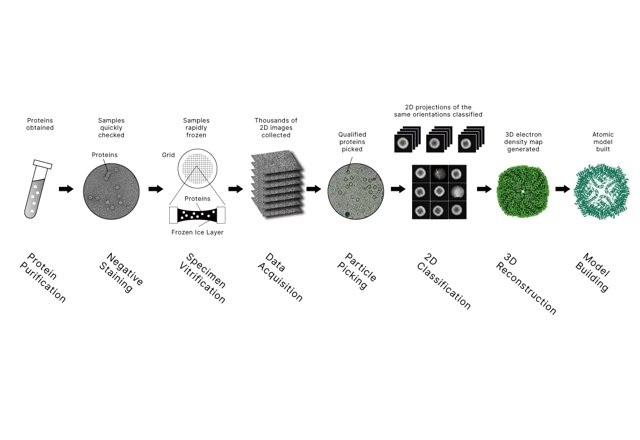
Cómo la crio-EM captura la dinámica de las proteínas
2025-07-30

Por qué el control de calidad del producto CAR-T debe utilizar un carroñero de ácidos nucleicos
2026-06-26

Detección de ARN de baja abundancia: Cómo una plataforma de imagen en gel de ARN de alta sensibilidad captura bandas débiles
2026-06-23
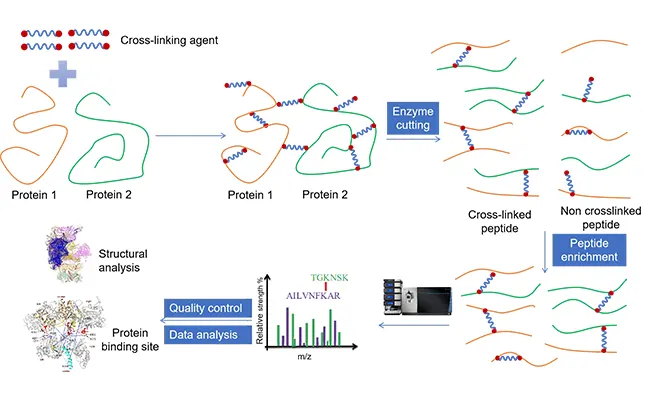
Espectrometría de masas de reticulación: preguntas frecuentes y soluciones probadas
2026-01-15La espectrometría de masas de reticulación ha madurado rápidamente, pasando de ser una técnica especializada a una piedra angular de la proteómica estructural y el mapeo de interactomas. Pioneros pioneros, como Ruedi Aebersold, Juri Rappsilber, Andrea Sinz y colaboradores, demostraron cómo XL-MS puede validar modelos crio-EM, resolver la organización de grandes máquinas macromoleculares y exponer contactos fugaces entre proteínas y que rara vez sobreviven a la purificación. Aprovechando este impulso, nuestro ecosistema integrado de servicios, instrumentos y consumibles ofrece flujos de trabajo reproducibles y de alto rendimiento para la espectrometría de masas de reticulación cruzada —desde la planificación de muestras hasta la interpretación de datos— diseñados para satisfacer las demandas del descubrimiento moderno. La espectrometría de masas de reticulación cruzada (XL-MS) ha evolucionado hasta convertirse en una herramienta fundamental para la proteómica estructural y el análisis de interectomas. Al capturar las restricciones espaciales entre residuos, XL-MS complementa la crio-EM y la cristalografía y revela interacciones transitorias proteína-proteína que a menudo escapan a los métodos basados en la purificación.
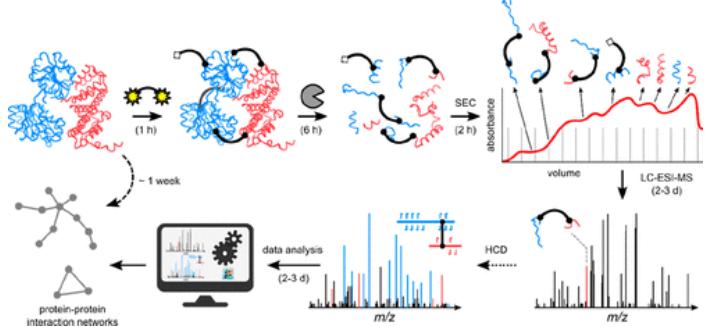
(Espectrometría de masas de reticulación cruzada para investigar conformaciones de proteínas
e Interacciones proteína-proteína - Un método para todas las estaciones)
Qué captura realmente la espectrometría de masas de reticulación
La espectrometría de masas de reticulación cruzada (también llamada reticulación química acoplada a espectrometría de masas, CL-MS o XL-MS) utiliza reactivos bifuncionales para unir covalentemente aminoácidos que se encuentran dentro de una ventana espacial definida. Tras la reticulación, la digestión enzimática dirigida genera una mezcla de péptidos lineales y entrecruzados. La espectrometría de masas detecta e identifica estas especies entrecruzadas, traduciéndolas en restricciones de distancia, socios de interacción e información específica del sitio que puede introducirse en modelos estructurales. Debido a que XL-MS proporciona restricciones derivadas de la proteómica, complementa la criomicroscopía electrónica y la cristalografía de rayos X aterrizando densidades ambiguas y refinando interfaces.
Más allá de su poder integrador, la espectrometría de masas de reticulación cruzada ofrece ventajas prácticas: escala a un alto rendimiento, soporta aplicaciones intracelulares y no requiere etiquetas de fusión genética ni marcaje químico especial. De manera crucial, el enlace cruzado covalente "congela" los contactos débiles o transitorios, haciendo de XL-MS un socio potente para la purificación de afinidad o la MS nativa cuando el objetivo es revelar interacciones que de otro modo se disiparían.
Por qué la adopción se retrasa: puntos de dolor persistentes
Incluso con beneficios claros, varios obstáculos siguen impidiendo el despliegue rutinario:
-Enriquecimiento y detectabilidad. Los péptidos reticulados suelen ser raros en comparación con los lineales. Sin un enriquecimiento o fraccionamiento cuidadosos, las tasas de identificación bajan y la cobertura de los sitios enlazables se vuelve irregular.
-Puntuación y validación complejas. Las masas precursoras compuestas y los patrones de fragmentación complejos complican la interpretación del espectro. El control de la tasa de descubrimiento de falsos (FDR) para pares encruzados requiere una puntuación consciente del sitio y umbrales cuidadosamente ajustados, o pueden pasar por alto enlaces espurios.
-Optimización química. La elección y la dosis del entrecruzado importan. La sobre-reticulación puede alterar las arquitecturas nativas; La subreticulación produce mapas dispersos. La reticulación en la célula añade complejidad matricial que puede reducir la recuperación de péptidos.
-Diseño de métodos de instrumentos. La sensibilidad debe equilibrarse con la calidad de fragmentación. Si solo usas HCD, la cobertura de fragmentos puede ser desigual; ETD/EThcD necesita calibración para asegurar que los iones útiles se recuperen para muchos pares de péptidos diferentes.
-Software y estándares fragmentados. Las herramientas heterogéneas y los formatos de informes dificultan comparar resultados entre proyectos o fusionar los resultados de XL-MS con bases de datos estructurales para modelado integrativo.
Preguntas frecuentes prácticas y soluciones probadas para la espectrometría de masas de reticulación cruzada
- ¿Cómo debería elegir una cruz?-¿Enlazador para mi sistema?
• Ajustar la longitud del espaciador a las distancias de interacción anticipadas y elegir grupos reactivos alineados con residuos dominantes (las químicas dirigidas a la lisina son comunes).
• Ajustar la concentración y duración de la reacción del reticulador en piloto para evitar el sobrecruzamiento.
•En matrices complejas, elegir químicas con reactividad bien caracterizada y protocolos de temple validados.
- ¿Cómo puedo mejorar la recuperación de la cruz?-¿Péptidos enlazados?
• Emplear flujos de trabajo de digestión secuenciales (por ejemplo, Lys-C y luego tripsina) para ampliar los límites de los péptidos y disminuir las escisiones perdidas.
• Utilizar enriquecimiento a nivel peptídico junto con fraccionamiento ortogonal para enfocar especies entrecruzadas por delante de LC-MS.
- Que LC-¿La configuración de MS aumenta las tasas de identificación? (Ningún método LC-MS único aplica para todos los péptidos reticulados. La optimización de métodos debería considerarse parte del flujo de trabajo XL-MS, no una configuración puntual.)
• Adquirir MS/MS de alta resolución y ajustar las energías de colisión para equilibrar la fragmentación de la columna vertebral y los entrecruzamientos.
• Considerar la fragmentación mixta (HCD suplementado con ETD o EThcD) para una mayor cobertura de secuencias de pares entrecruzados.
- ¿Cómo controlo los falsos descubrimientos?
• Aplicar enfoques de señuelo objetivo adaptados a búsquedas cruzadas y aplicar filtros de puntuación delta, a nivel de sitio y tipo enlace.
• Validar enlaces críticos entre réplicas biológicas y técnicas y verificar con ensayos bioquímicos criogenicos-EM o ortogonales.
- ¿Puede la espectrometría de masas de reticulación cruzada capturar interacciones transitorias en células vivas?
•Sí: la reticulación realizada dentro de las celdas preserva las interacciones fugaces. Optimizar el temple y la lisis para asegurar la integridad del aducto, y utilizar el enriquecimiento para gestionar los efectos de la matriz antes de la EM.
- ¿Cómo integro XL?-Esclerosis múltiple con criogenia-EM o X-¿raya?
• Proporcionar límites de distancia específicos del linker y métricas de confianza. Utiliza restricciones XL-MS para orientar dominios, confirmar interfaces y marcar regiones discordantes para el refinamiento del modelo.
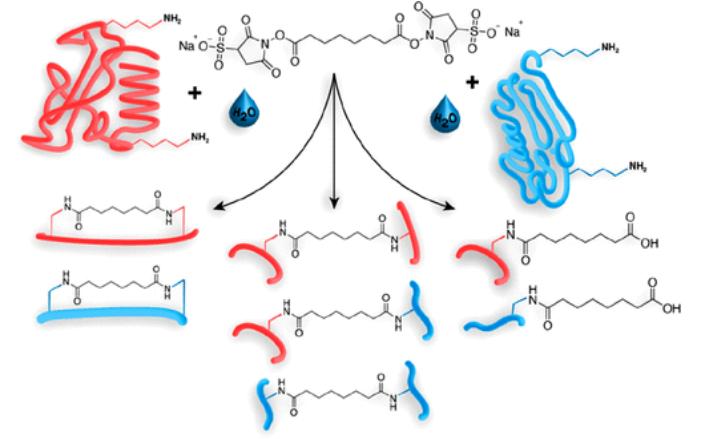
(Reacción de ésteres NHS reactivos a aminas, ejemplificada para BS3 (panel superior).
Los productos de reacción con proteínas incluyen enlaces cruzados intrapeptídicos (Tipo 1; Izquierda),
Enlaces cruzados interpeptídicos (Tipo 2; Medio),
y enlaces cruzados "callejón sin salida" o "mono-enlaces" (Tipo 0; Correcto))
Una pila integrada de servicios y productos que acelera XL-MS
Ofrecemos un Servicio XL-MS de extremo a extremo, que cubre el diseño experimental, la optimización química, la preparación de muestras, la adquisición de LC-MS e interpretación de datos. Este enfoque integrado minimiza el ensayo y error, reduce los descubrimientos falsos y acorta el camino desde los experimentos piloto hasta los resultados publicables. Los clientes pueden enviar muestras pre-encruzadas o co-diseñar el flujo de trabajo con nuestro equipo desde el principio.
- Por qué nuestras soluciones genómicas y equipos de laboratorio fortalecen XL-MS?
XL-MS se beneficia de una preparación estable de muestras aguas arriba y una detección robusta aguas abajo. Ofrecemos instrumentos avanzados, reactivos de alta calidad y consumibles que soportan la espectrometría de masas de reticulación cruzada y las canalizaciones ómicas relacionadas en entornos académicos, clínicos e industriales. Guiados por la eficiencia y la precisión, nuestros flujos de trabajo apoyan estudios de alto rendimiento con una calidad de datos consistente, condiciones ideales para una interpretación segura de espectros de péptidos reticulados.
- Perfil de interacción ChIP-Seq y cromatina
Cuando los proyectos analizan los contactos entre proteínas y cromatinas junto con la espectrometría de masas de reticulación, ChIP-seq añade contexto genómico específico del locus. Al mapear factores de transcripción y sitios de unión de histonas a nivel genómico, ChIP-seq ayuda a conectar las restricciones XL-MS con los paisajes regulatorios funcionales, reforzando las conclusiones mecanicistas y guiando hipótesis para experimentos posteriores.
- Genómica y Apoyo a la Preparación de Muestras
Longlight se centra en biología molecular y diagnóstico molecular con un portafolio de instrumentos y reactivos relacionados con NGS, incluyendo soluciones de ultrasonicación enfocadas para la preparación precisa de bibliotecas. Estas capacidades estabilizan la calidad de la muestra aguas arriba —un factor que a menudo se pasa por alto del éxito— de modo que la espectrometría de masas de encruzamiento produce resultados interpretables y reproducibles entre lotes y proyectos.
- Consumibles y kits que reducen la variabilidad
Suministramos geles de agarosa prefabricados, carroñeros de ácidos nucleicos, tubos de Qubit, kits de extracción de ácidos nucleicos y kits de preparación de bibliotecas. El uso de consumibles comunes y procedimientos operativos estándar estrictos reduce la variabilidad diaria, ayudando a los equipos a ofrecer un rendimiento estable de XL-MS a través de mediciones repetidas e integraciones multi-ómicas.
- Recomendaciones simplificadas para evitar errores comunes
• Ejecutar titulaciones piloto para ajustar la concentración de reactivos y el tiempo de reacción por matriz.
• Utilizar flujos de trabajo estandarizados de proteasas y enriquecimiento a nivel de péptido para aumentar el rendimiento de péptidos reticulados.
• Mantener registradas las versiones de los métodos LC - MS y, cuando sea apropiado, favorecer fragmentación híbrida HCD/ETD o EThcD de alta resolución.
• Establecer filtros de puntuación estricta, puntaje delta y a nivel de sitio, y reproducir enlaces prioritarios en réplicas técnicas y biológicas independientes.
• Combinar espectrometría de masas de reticulación cruzada con crio-EM o rayos X y revelar límites de distancia y métricas de confianza específicos del enlace para modelado integrativo.
De piloto a escala: tu siguiente paso
¿Planificar la espectrometría de masas de entrecruzamiento para mapeo de interacciones proteicas, validación de estructuras o modelado integrativo? Colabora con nuestros expertos para diseñar un flujo de trabajo fiable y de alto rendimiento, optimizado para tus objetivos. Asesoraremos sobre la selección de los entrecruzados, organizaremos la presentación de muestras y realizaremos digestión, enriquecimiento, adquisición de EM y análisis de datos. Espera un informe conciso con información accionable. Colabora con nosotros para reducir riesgos, solucionar cuellos de botella comunes y elevar XL-MS de pilotos tempranos a un descubrimiento fiable y cotidiano.